Según la definición de la ILAE de 2014 (Fisher et al. 2014), la epilepsia es una enfermedad debida a una alteración del normofuncionamiento del sistema nervioso central, en particular de la corteza cerebral, que se caracteriza por la aparición de síntomas de diverso tipo (motor, sensitivo, psíquico) de forma transitoria y repetitiva (paroxística), involuntaria, que afecta al normofuncionamiento y la calidad de vida del individuo.
Es necesario (pero no suficiente) que un individuo haya presentado al menos 1 crisis epiléptica para el diagnóstico de epilepsia. Además, se exige la presencia de un riesgo mayor al normal de presentar un nuevo episodio para que se pueda realizar el diagnóstico de epilepsia. Este riesgo mayor al normal puede ser demostrado de varias formas, según la definición operacional de la ILAE de 2017 (Fisher 2017):
- La presentación de al menos 2 crisis epilépticas no provocadas (o reflejas), separadas más de 24h.
- La presentación de al menos 1 crisis epiléptica no provocada (o refleja), y una probabilidad similar al riesgo de recurrencia general después de haber presentado 2 crisis epilépticas no provocadas (al menos de un 60% en los próximos 10 años), por la presencia de lesiones cerebrales o otros biomarcadores que así lo sugieran.
- El diagnóstico de un síndrome epiléptico.
El término epilepsia no tiene una implicación etiológica, y pueden existir distintas causas que la produzcan.
Se considera que una epilepsia se ha curado cuando se trata de una epilepsia edad-dependiente y el individuo ya no tiene una edad compatible con el síndrome, o cuando han pasado al menos 10 años sin crisis, y de éstos los últimos 5 años sin tomar medicación antiepiléptica.
Clasificación de los tipos de crisis y de las epilepsias infantiles.
La clasificación de las epilepsias es un tema de gran polémica que ha sufrido varias modificaciones en los últimos años. En la última revisión de la ILAE de 2017 se revisó la clasificación y terminología de las epilepsias (Scheffer et al. 2017).
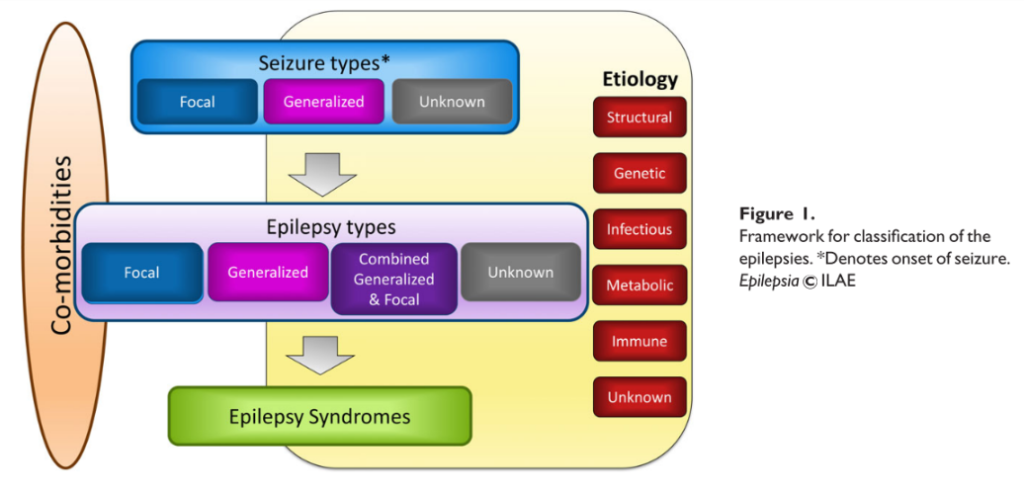
Ilustración 6. Estructura general del sistema de clasificaicón de las epilepsias de la ILAE.
- Tipo de crisis epiléptica (primer nivel de diagnóstico). El punto de partida del marco de clasificación de las epilepsias es el tipo de crisis, una vez hemos realizado el diagnóstico de crisis epiléptica y se han diferenciado de éstos los episodios compatibles con trastornos no epilépticos. En algunas situaciones la clasificación por el tipo de crisis podría ser el nivel máximo de diagnóstico, por no existir información suficiente, imposibilidad de acceso a pruebas diagnósticas, o número insuficiente de episodios. Se pueden clasificar en función de su forma de inicio:
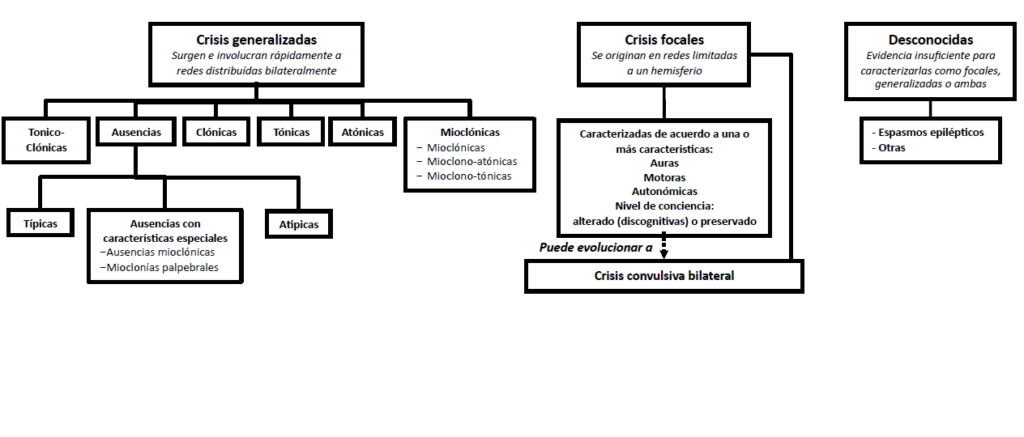
Ilustración 7. Clasificación de los tipos de crisis según la ILAE.
- De inicio focal. La principal característica de una crisis focal es que en su inicio afecta exclusivamente a un hemisferio cerebral, y por lo tanto suele presentar síntomas productivos unilaterales relacionados con el área cerebral en la que se produce la descarga epiléptica. Las crisis focales pueden subclasificarse en función de los síntomas que presentan a su inicio en:
- Motoras
- Sensitivas
- Autonómicas
- Nivel de consciencia: Alterado o preservado.
- De inicio generalizado. Desde su inicio afectan a ambos hemisferios simultáneamente, lo cual significa que cumplen al menos 1 de los siguientes: o producen afectación de la consciencia desde su inicio, no habiendo memoria alguna del episodio, y/o en caso de existir síntomas motores, éstos son bilaterales. Existen múltiples subtipos de crisis generalizadas en función de los síntomas motores que aparecen:
- Las ausencias son un tipo de crisis generalizada caracterizada en su forma típica principalmente por presentar desconexión del medio, habitualmente de inicio súbito, de breve duración. Son particularmente frecuentes en la infancia
- Mioclónicas. Las crisis mioclónicas se caracterizan por presentar preservación del nivel de consciencia (serían las únicas crisis generalizadas con preservación de ésta), acompañada por episodios de movimientos súbitos, aislados, asíncronos, descoordinados, bilaterales, de las extremidades (tipo sacudida).
- Tonico-clónicas. Se caracterizan por la afectación del nivel de consciencia acompañada de una contracción tónica de todo el cuerpo seguida o no de movimientos de contracción-relajación rítmicos, bilaterales y síncronos.
- Atónicas. Las crisis atónicas se caracterizan por episodios súbitos de pérdida del tono muscular.
- De inicio desconocido. Como los espasmos epilépticos infantiles o del lactante.
- Tipo de epilepsia (segundo nivel de diagnóstico). También puede ser el nivel último de diagnóstico si el clínico no posee información suficiente como para realizar un diagnóstico de síndrome epiléptico.
- Epilepsia focal. Las epilepsias focales incluyen los trastornos unifocales o multifocales, así como las crisis que afectan un único hemisferio cerebral. (Panayiotopoulos 2005).
- Crisis del lóbulo temporal
- Crisis del lóbulo frontal
- Crisis del lóbulo parietal
- Crisis del lóbulo occipital
- Epilepsia generalizada. Para su diagnóstico el paciente debe presentar actividad generalizada de punta-onda en el EEG. Pueden tener distintos tipos de crisis, como ausencias, mioclonicas, tónicas, atónicas y tonicoclónicas
- Epilepsia combinada focal y generalizada. Existen pacientes que combinan características de los dos grupos anteriores, como por ejemplo los pacientes con síndrome de Dravet o síndrome de Lennox-Gastaut.
- Desconocida. En dicha categoría se incluyen aquellos pacientes con un diagnóstico inequívoco de epilepsia, pero que no es posible clasificar en ninguno de los grupos anteriores.
- Epilepsia focal. Las epilepsias focales incluyen los trastornos unifocales o multifocales, así como las crisis que afectan un único hemisferio cerebral. (Panayiotopoulos 2005).
- Síndrome epiléptico (tercer nivel diagnóstico): Un síndrome epiléptico se define como un conjunto de fenómenos que incluyen tipo de crisis, EEG y neuroimagen, que tienden a producirse conjuntamente. Suelen tener una presentación edad-dependiente (edad de inicio y remisión típicas), desencadenantes específicos, ritmo circadiano y en muchas ocasiones un mismo pronóstico. Pueden presentar asimismo comorbilidades específicas como discapacidad intelectual y trastornos psiquiátricos. También pueden tener implicaciones etiológicas, pronósticas y terapéuticas, aunque los síndromes epilépticos no tienen una correlación uno a uno con los diagnósticos etiológicos. Existen múltiples síndromes bien conocidos, como la epilepsia de ausencias infantil, el síndrome de West, o el síndrome de Dravet. Este marco de clasificación es utilizado para toda la población de pacientes epilépticos, tanto adultos como niños, pero existen varias circunstancias particulares de la edad pediátrica que merece la pena reseñar. La mayor parte de los síndromes epilépticos de la infancia son edad-dependientes, por lo que dentro de los síndromes epilépticos podemos realizar una subclasificación en función de la edad de inicio.
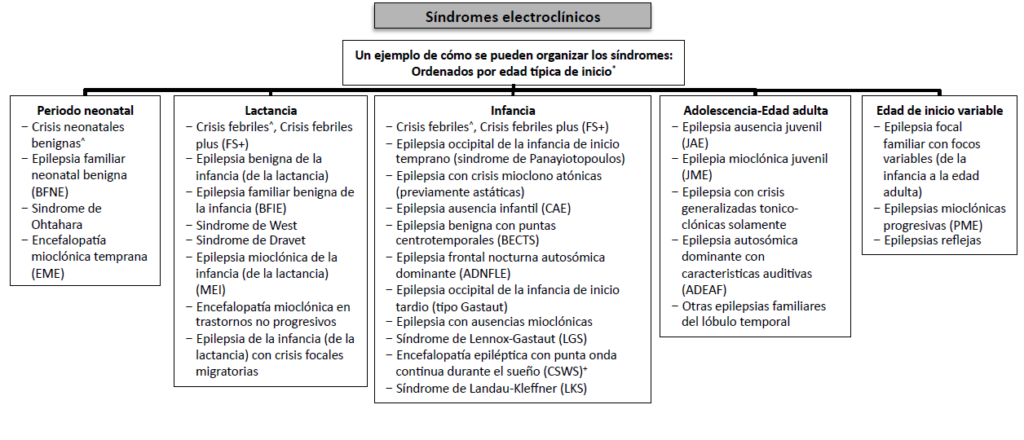
Ilustración 8. Clasificación de los síndromes epilépticos según la ILAE.[/vc_column_text][/vc_column][/vc_row]


